Research
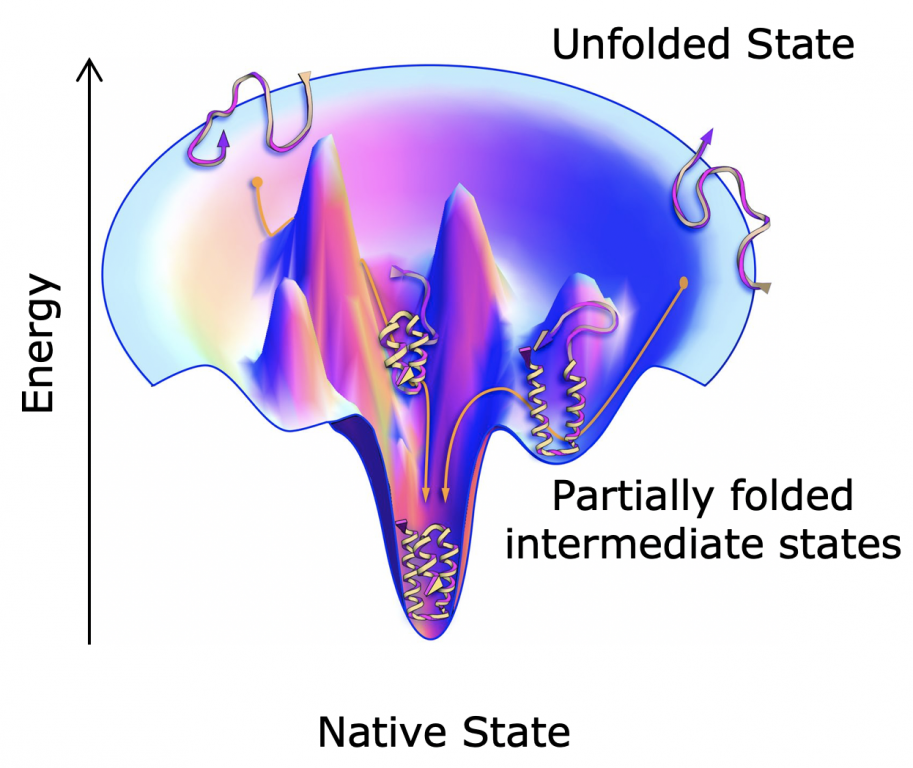
Folding through the energy landscape
We are interested in understanding the structural and dynamic information encoded in the linear amino acid sequence of proteins and how environmental factors can modulate this relationship. The amino acid sequence encodes the protein structure and the various conformations available to the polypeptide chain, and we describe the conformational space of a protein as an energy landscape. Our lab uses a combination of biophysical, structural, biochemical, and computational techniques to understand how changing a protein sequence and environment can affect protein folding, binding, degradation, allostery, and signaling.
Single-molecule protein studies
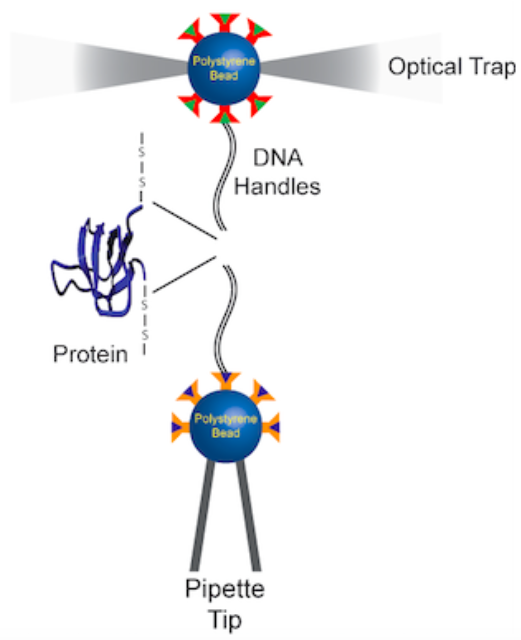
Force spectroscopy with optical tweezers is a valuable technique that gives insight into the protein folding process at a single-molecule level. This technique can be used to understand the effect of mechanical force on the energy landscape of protein folding and unfolding, and to detect rare pathways and intermediates.
Two beads are attached to a molecule. One bead is tethered to a micropipette, the other to a laser trap, and force is applied. This technique can provide information on kinetics and thermodynamics of conformational changes measured by extension changes.
We have also combined MD simulations with single-molecule experiments to explore the complexity of energy landscapes and investigate alternative folding pathways.
Protein folding in the cell
The cellular environment is often vastly different from in vitro conditions. The presence of other macromolecules and post-translational modifications play roles in protein folding and dynamics that we are only beginning to understand. We are currently focusing our efforts on studying co-translational folding and protein ubiquitination, in addition to exploring how protein dynamics play a role in signal transduction pathways.
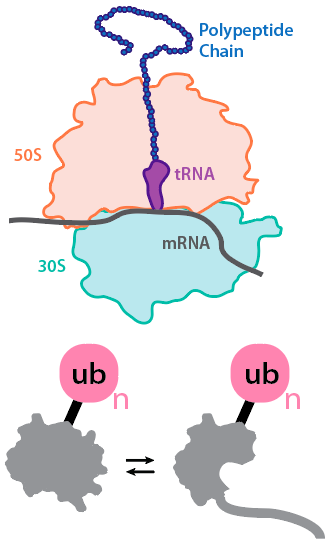
All proteins have the opportunity to fold co-translationally, raising questions regarding the structure and dynamics of the nascent polypeptide as it emerges from the ribosome. We are using proteolysis, fluorescence, mass spectrometry, and single-molecule approaches to understand how the ribosome and the vectorial nature of translation affect folding in vitro and in vivo.
Protein substrates in the cell are also often modified post-translationally via ubiquitination, phosphorylation, and other PTMs. We are developing methodologies for studying how ubiquitination affects the local and global stabilities of substrate proteins and how this relates to degradation by the proteasome.
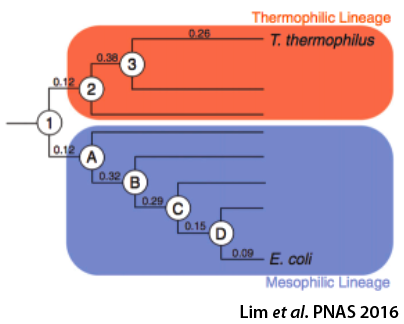
Effects of sequence variation on energy landscapes
Protein sequences change over evolutionary time in response to evolutionary pressures and natural drift. As the sequence changes, so does the energy landscape and its associated biophysical parameters. We are using Ancestral Sequence Reconstruction (ASR) methods to probe how sequence space and energy landscapes have changed over time. Both ASR and the consensus method have been shown to result in proteins that are more stable than the starting extant sequences. We are applying both methods to the RNase H family to explore the differences between the resulting proteins.